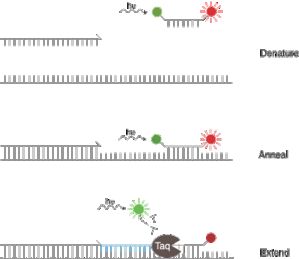
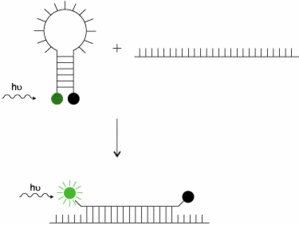
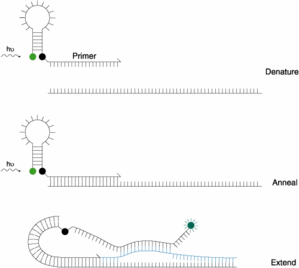
Now that you have seen a typical qPCR system using a TaqMan probe, let is go to the next step : Duplex.
First of all : why is it better to do a duplex PCR, that is to say having 2 PCR system in the same well with 2 TaqMan probes?
I could answer you briefly without any clear explanation talking about identification, inhibition, control and genotyping. But you will be surely a bit lost and you will not know more about why and how...
One of the main interest for duplex qPCR is : inhibition control
Indeed, for a trivial reason, PCR is an enzymatic reaction prone to inhibition from different compounds. Inhibition has also a major role in the result given. When a positive result is obtained, there is not a lot of question about it, it is taken like that.
On the other hand, when result is negative, does it come from that there is definitely not any target in the sample (true negative) or because the sample is inhibited and PCR does not work well (false negative). How to know?
Using inhibition control is the answer.
This one is composed by a primer and probe system but also by a target DNA. In order to distinguish both in the same well, a difference has to be exploited. The basis fot that is to use a grafted probe with a really different fluorophore from the target one.
Following combination is obtained so :
target DNA
target primers
target probe grafted by FAM for example.
control DNA
control specific primers
control specific probe grafted with a different probe than the target one
Among available fluorophores, the range is wide and can be seen for example on provider website like Biosearch or Eurogentec.
The most used ones are FAM (again) and VIC even if it is not the best combination for optimum use, but it is linked to PCR history when these two molecules were the almost only ones on the market, doing good job for duplex. Today, many other exist...
Let is say that we will use FAM and VIC fluorophores for our duplex example.
It is very important to check fluorophores overlapping for excitation and emission spectra, because it can have a very big impact on detection signal if not well managed. Obviously, FAM and VIC fluorophores have overlapping spectra that has to be compensated then.
These closed spectra induce interferences between the two signals as we would like to discriminate them.
FAM signal (here in green, and one of the strongest emiting fluorophores on the market) overlap the VIC one (in yellow) in a pretty important part. Compared to TAMRA (a lighter overlapping) and Cy5 (no overlap at all), VIC is strongly interfered by FAM. Both signals will be biased and will give bad results for quantification.
To get rid of this problem, a color compensation has to be made. It is a little amplification to do one for all to help the qPCR device to discriminate between both signals. Normally, device supplier can help you if the program is not mentioned in the device notice.
A compensation run comprises a complete mix with two fluorophores and target nucleic acid, but also all the fluorophores in a single detection. When run is done, it will generate a program helping the software device to better discriminate the signals for any detection then.
VIC signal is pretty often the Inhibition Control (IC) signal. And its signal has to be always the same for the Ct. If IC Ct for any sample is the same as the control one, the sample is not considered as inhibited. If the sample Ct IC undergo a Ct shift of at least 3 Ct (1 log), it means that sample is inhibited and that the quantification or detection made is wrong.
If a partial inhibition, IC signal will be shifted and curves flattened, but still positive.
If a complete inhibition, IC signal is not present anymore.
Here a synthesis of results that can be obtained :
| Target | Control | PCR résult | Comments |
| + | + | Positive that can be quantified | True Positive |
| + | - | Positive with partial inhibition | Partial Inhibition |
| - | + | Negative | True negative |
| - | - | Inhibited | Total Inhibition |
In the same well, a detection and quantification can be made for a sample but also insuring the results because of inhibition control avoiding true negative.
As told before, many qPCR duplex are made using FAM-VIC, but need color compensation, whereas many other conbination exists working at least the same with less proble to set up (like Cy5-VIC)
In recent years, Quenchers have been improved to replace useful but fluorescent TAMRA one. New quenchers are non-fluorescent and can also help hybridization of target probe like MGB solution from Life Technologies/Applied Biosystem. Multiplex are easier to set up because no backgournd signal coming from emiting quencher like TAMRA.
Most used ones are :
- BHQ (Black Hole Quenchers) I, II et III , depending on the reporter used
- DDQ (Eclipse Dark Quenchers) : similar to BHQ
- MGB (Minor Groove Binder) : molecule improving probe affinity to DNA and comprising also a NFQ (non fluorescent quencher) developed by ABI.
It has to be noticed that using a duplex need quite often working on qPCR parameters to optimize it like IC DNA to use, primers and probe concentrations, hybridization T°, MgCl2 concentration or adjuvant adding.
More details in another post, but also few words on triplex, fluorophores and tips from my PCR experience.
Have good amplifications....